Кройцфелд-Якоб (CJD) — прионно заболяване: дефиниция, причини, симптоми
Кройцфелд-Якоб (CJD) — прионно дегенеративно заболяване: симптоми, причини, диагноза и рискове. Научете как се проявява и какво означава за вашето здраве.
Болестта на Кройцфелд-Якоб (произнася се KROITS-felt YAH-kohb) или CJD е неврологично заболяване. То е дегенеративно (влошава се с течение на времето), не може да бъде излекувано и винаги води до смърт. Понякога CJD се нарича човешка форма на "болестта на лудата крава" (спонгиформна енцефалопатия по говедата, или BSE). Всъщност СЕГ е причина за един рядък вид болест на Кройцфелд-Якоб; двете не са едно и също заболяване.
CJD се причинява от инфекциозенагент, наречен прион. Прионите са протеини, които са сгънати неправилно. Прионите създават свои копия, като променят правилно сгънатите протеини в неправилно сгънати форми. CJD причинява много бързо увреждане на мозъчната тъкан. Тъй като болестта разрушава мозъка, в него се появяват дупки. Структурата на мозъка се променя и той става като кухненска гъба.
Галерия с изображения
6 Изображения




Какво представлява прионът и защо е опасен
Прионите са белтъци, които могат да променят формата си и да индуцират еднаква промяна в нормалните мозъчни протеини. Това води до натрупване на неправилно сгънати протеини, до увреждане на нервните клетки и до образуване на характерни "дупки" в мозъчната тъкан (спонгиформна дегенерация). Прионните заболявания са редки, но прогресират бързо и са фатални.
Видове Кройцфелд-Якоб
- Спорадичен CJD — най-честата форма (около 85–90% от случаите). Възниква спонтанно без ясна причина, обикновено при хора на средна или напреднала възраст.
- Наследствен (генетичен) CJD — причинен от мутации в гена PRNP и предаващ се по рода; клиничната картина може да варира.
- Иатрогенен CJD — пренесен по медицински път, например чрез замърсени неврохирургични инструменти, кориални транспланти, трансфузии с използвана кръвна плазма в миналото или прилагане на хормони (хорашен растежен хормон) от замърсени източници.
- Вариантен CJD (vCJD) — свързан с експозиция на BSE (болестта на "лудата крава"); засяга по-млади пациенти и често започва с поведенчески и психиатрични симптоми.
Причини и механизъм
Причинителят е прион — неправилно сгънат белтък, който може да преобразува нормалните мозъчни протеини в патологична форма. Това води до каскада на натрупване, възпаление и загуба на неврони. В наследствените случаи промяната е генетично обусловена (мутации в PRNP), а при иатрогенните случаи прионът се пренася от заразени материали.
Симптоми
Началото и комбинацията от симптоми могат да варират, но обикновено заболяването прогресира бързо. Често срещани прояви:
- Бързо влошаваща се деменция — загуба на памет и мислене;
- Промени в поведението, безпокойство или депресия;
- Неврологични симптоми: координационни нарушения (атаксия), мускулни потрепвания (миоклонус), слабост, нарушение в речта и преглъщането;
- Зрителни нарушения и слепота (по-често при някои подтипове);
- При vCJD — начални психиатрични симптоми (депресия, тревожност), последвани от неврологични нарушения.
Диагноза
Диагнозата се поставя чрез комбинация от клинична оценка и специфични изследвания:
- Неврологичен преглед и проследяване на бързото прогресиране на симптомите;
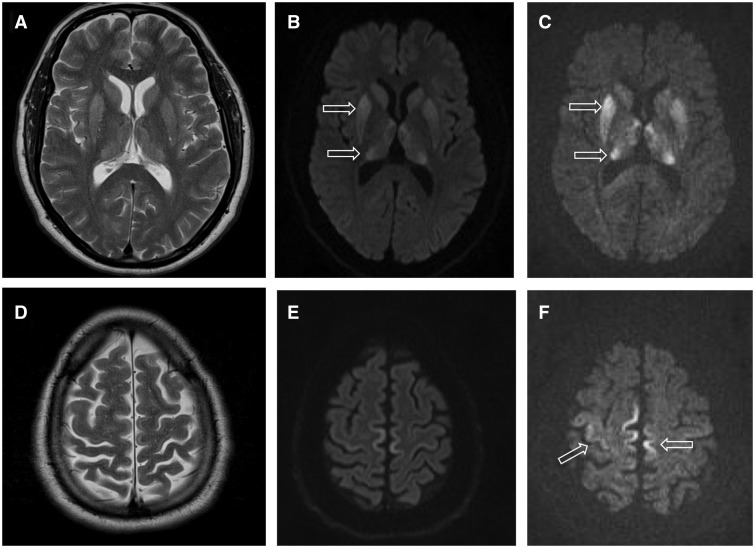
- Ядрено-магнитен резонанс (MRI) — характерни промени в определени части на мозъка (например "pulvinar sign" при vCJD и други специфични находки при sCJD);
- Електроенцефалография (EEG) — може да покаже периодични остри вълни при някои пациенти;
- Анализ на гръбначно-мозъчната течност (CSF) — белтъци като 14-3-3 или повишено общо tau могат да подпомогнат диагнозата; най-съвременните методи като RT-QuIC имат висока чувствителност и специфичност за откриване на прионна активност;
- Биопсия на мозъка или аутопсия — потвърждава диагнозата чрез директно идентифициране на прионните промени, но се използва рядко при живи пациенти поради рисковете и сложността.
Лечение и прогноза
Към момента няма ефективно лечение, което да излекува CJD или да спре прогресията на заболяването. Поддържащите мерки са основата на грижата:
- Подобряване на комфорта и симптоматично лечение (обезболяване, лечение на тревожност и депресия, контрол на миоклонуса);
- Поддържаща грижа — помощ при хранене, хидратация и ежедневни нужди; често се налага хосписна грижа;
- Палиативни и мултидисциплинарни подходи за осигуряване на качество на живот при терминалния стадий.
Прогнозата е тежка: повечето пациенти с спорадичен CJD умират в рамките на месеци до около година след появата на симптомите. Вариантният и наследственият типове могат да имат различно времево протичане, но са също фатални.
Превенция и контрол
- Стриктни мерки за контрол на инфекцията в болнична среда — правилна дезинфекция и стерилизация на медицински инструменти;
- Ограничаване на използването на човешки тъкани и препарати с риск от прионно замърсяване; в миналото са предприемани забрани и тестове за кръвни и трансплантационни процедури за намаляване на риска;
- Надзор и контрол в животновъдството за превенция на BSE и мерки за безопасност в хранителната верига — това пряко намалява риска от vCJD;
- Информиране и образоване на медици и обществеността за ранните признаци и рисковите процедури.
Епидемиология
CJD е много рядко заболяване — спорадичният тип се среща приблизително при 1–2 случая на милион души годишно. vCJD е значително по-рядък и се появи в по-голяма степен в области, където бе епидемия от BSE сред говедата през 1980–1990-те години.
Кога да потърсите лекар
Ако забележите бързо влошаване на паметта, сериозни промени в мисленето или необясними неврологични симптоми, потърсете спешно медицинска помощ. Ранната оценка от невролог може да насочи към правилни диагностични тестове и адекватна поддържаща грижа.
Ключови точки
- CJD е редко, бързо прогресиращо и фатално прионно заболяване.
- Причинява се от неправилно сгънати прионни белтъци; има спорадична, наследствена, иатрогенна и вариантна форма.
- Диагнозата се поставя чрез клинична оценка, образни и лабораторни изследвания; потвърждение често става постмортем.
- Няма лечение, което да излекува болестта — грижите са симптоматични и палиативни.
Видове и причини за CJD
Видовете CJD включват:
- вариант (vCJD):
Този вид CJD може да бъде причинен от консумация на храна, в която има приони, като например месо от крави, болни от СЕГ ("болест на лудата крава"). Това обаче е много рядка причина за CJD.
- спорадична (sCJD):
Това е най-разпространеният тип CJD. 85 % от случаите на CJD са спорадични CJD. Никой не знае какво причинява sCJD; изглежда, че това се случва случайно.
- фамилна (fCJD):
Повечето от останалите 15 % от случаите на CJD са фамилни CJD. Това е форма на CJD, която се среща в семействата.
- ятрогенен:
Тази форма на CJD обикновено се причинява от медицинска процедура, при която човек получава кръв или тъкан от болен от CJD. Например, човек може да получи ятрогенна CJD, ако му бъде прелята кръв или трансплантирана роговица от човек, който има CJD.
Признаци и симптоми
Първият симптом на CJD е деменция, която се влошава много бързо. деменцията причинява загуба на паметта, промени в личността и халюцинации.
Други често срещани психични симптоми включват:
- Тревожност
- Депресия
- Параноя
- Обсесивно-компулсивни симптоми
- Психоза
Физическите симптоми на CJD често включват:
- Проблеми с говора
- Отривисти движения (миоклонус)
- Проблеми с равновесието (атаксия)
- Проблеми с ходенето
- Разклащане или скованост
- Проблеми със зрението
- Проблеми с преглъщането, които могат да затруднят или да направят невъзможно храненето.
- Проблеми с кашлицата, които могат да причинят пневмония
- Движения, които пациентът не може да контролира (дискинезия)
Повечето хора с CJD умират в рамките на шест месеца след появата на първите симптоми. Често те умират от пневмония, причинена от проблеми с кашлицата. Около 15% от пациентите преживяват две или повече години. Някои пациенти живеят 4-5 години с предимно психични симптоми, докато болестта се влоши и предизвика повече физически симптоми. След като това се случи, хората обикновено умират в рамките на една година.
Симптомите на CJD се дължат на смъртта на все повече нервни клетки в мозъка. Когато учените разглеждат под микроскоп мозъчна тъкан от пациент с CJD, те могат да видят множество малки дупчици, в които са загинали цели области от нервни клетки.
Диагноза
Лекарите могат да заподозрат CJD, когато човек има определени симптоми. Например деменцията обикновено се влошава бавно. Деменция, която се влошава много бързо, е необичайна. Заедно със симптоми като отривисти движения, тези симптоми могат да насочат към възможна CJD.
След това могат да се направят тестове, за да се установи дали лицето има CJD. Тези изследвания включват:
- Електроенцефалография (ЕЕГ): Това изследване показва електрическата активност в мозъка. Лекарят често може да забележи промени в ЕЕГ, които са характерни за хората с CJD. Видът на промените, които се виждат при ЕЕГ, зависи от вида на CJD и от това, в каква фаза е заболяването.
- Лумбална пункция (спинална пункция): Това изследване позволява да се изследва гръбначно-мозъчната течност (течността, която заобикаля главния и гръбначния мозък), като се търси специфичен протеин ("протеин 14-3-3").
- ЯМР на мозъка: Изследване, при което с помощта на много силен магнит се правят снимки на мозъка.
- Биопсия: За да направи биопсия, хирургът използва игла, за да вземе малко парче тъкан от тялото, така че лекарите да могат да го разгледат под микроскоп. vCJD може да се диагностицира с биопсия на сливиците. За всички останали видове CJD биопсията на мозъка е единственият начин да се установи със сигурност дали човек има CJD. Въпреки това, тъй като биопсията на мозъка може да причини увреждане на мозъка, биопсия на мозъка обикновено не се прави, ако други изследвания вече са показали, че лицето вероятно има CJD.

Лечение
Към 2016 г. не съществува лечение, което да излекува CJD или дори да забави ефектите му. Провеждат се много експерименти, за да се намери лечение.
Днес единственото лечение на CJD са лекарства, които лекуват симптомите на болестта и помагат на пациентите да се чувстват по-добре. Например на пациентите, които имат припадъци, могат да се дават антиконвулсивни лекарства. Бензодиазепините могат да намалят честотата на мускулните потрепвания.
Пациентите могат да изберат и медицински процедури, които да им помогнат да се справят с лошите симптоми. Например, CJD може да причини толкова големи проблеми с преглъщането, че човек да не може да се храни. Някои хора с CJD избират да им бъде поставена тръба за хранене, когато вече не могат да се хранят. Това е тръба, която се вкарва в стомаха, така че специална течност може да се подава директно в стомаха, за да се осигури храна на човека.
Свързани страници
- Прион
- Прионно заболяване
- Терминално заболяване
Въпроси и отговори
В: Какво представлява болестта на Кройцфелд-Якоб?
О: Болестта на Кройцфелд-Якоб (CJD) е неврологично заболяване, което е дегенеративно, нелечимо и винаги с фатален край.
В: Има ли лечение за CJD?
О.: Не, няма лечение за CJD.
В: Защо понякога CJD се нарича човешка форма на "болестта на лудата крава"?
О: Понякога CJD се нарича човешка форма на "болестта на лудата крава", защото спонгиформната енцефалопатия по говедата (BSE), която е причина за един рядък вид CJD, е известна като "болест на лудата крава".
В: Каква е причината за CJD?
О: CJD се причинява от инфекциозен агент, наречен прион, който представлява неправилно сгънат протеин, който може да създава свои копия, като променя правилно сгънатите протеини в неправилно сгънати.
В: Какво се случва с мозъчната тъкан при CJD?
О: CJD причинява много бързо нездравословно увреждане на мозъчната тъкан, което води до появата на дупки в мозъка и промяна в структурата на мозъка, който става като кухненска гъба.
В: Същото заболяване като CJD ли е BSE?
О: Не, СЕГ не е същото заболяване като CJD; всъщност тя е причина за един рядък вид CJD.
В: Как прионите причиняват CJD?
О: Прионите причиняват CJD, като се сгъват неправилно и създават свои копия за сметка на правилно сгънатите протеини в мозъка. Това води до разрушаване на здрава мозъчна тъкан и до появата на характерните за болестта дупки.
Свързани статии
Автор
AlegsaOnline.com Кройцфелд-Якоб (CJD) — прионно заболяване: дефиниция, причини, симптоми Leandro Alegsa
URL: https://bg.alegsaonline.com/art/24152
Източници
- cdc.gov : "CJD (Creutzfeldt–Jakob Disease, Classic)"
- ncbi.nlm.nih.gov : "Creutzfeldt–Jakob disease: Transmissible spongiform encephalopathy; vCJD; CJD; Jacob-Creutzfeldt disease"
- bmj.com : "Bovine spongiform encephalopathy and variant Creutzfeldt–Jakob disease"
- doi.org : 10.1101/SQB.1996.061.01.052
- pubmed.ncbi.nlm.nih.gov : 9246478
- www3.interscience.wiley.com : "MM2-cortical-type sporadic Creutzfeldt–Jakob disease with early stage cerebral cortical pathology presenting with a rapidly progressive clinical course"
- doi.org : 10.1111/j.1440-1789.2008.00904.x
- pubmed.ncbi.nlm.nih.gov : 18410280
- who.int : who.int: "Fact sheets no 180: Variant Creutzfeldt-Jakob disease" Feb 2012 ed.
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- doi.org : 10.1056/NEJM198610163151605
- pubmed.ncbi.nlm.nih.gov : 3762620
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- merckmanuals.com : "Creutzfeldt–Jakob Disease (CJD)"
- ncbi.nlm.nih.gov : "Creutzfeldt-Jakob disease: Updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy"