Синдром на Патау (тризомия 13): причини, симптоми и риск
Синдром на Патау (тризомия 13): причини, симптоми и рискови фактори при бременност — диагноза, профилактика и подкрепа за семейства.
Синдромът на Патау, известен още като тризомия 13 или тризомия D, е проблем с хромозомите. Хората, които страдат от него, имат допълнително копие на хромозома 13. Обикновено това се дължи на проблем, възникнал по време на мейозата, но може да е резултат и от Робертсонова транслокация — често срещано пренареждане на хромозомите при хората. Рискът да се получи проблем по време на мейозата се увеличава, когато жените имат деца на по‑късна възраст. Средната възраст на майките, при които се раждат деца с този синдром, е около 31 години.
Това е най-рядката от трите често срещани тризомии. По-често срещани са синдромът на Даун и синдромът на Едуардс. Синдромът на Патау засяга приблизително едно на 25 000 живородени деца, като честотата варира и се увеличава с възрастта на майката.
Галерия с изображения
4 Изображения


Причини и генетични форми
Основната причина е наличието на допълнително копие на хромозома 13. Има три основни генетични форми:
- Пълна тризомия 13 — повечето клетки в организма имат три копия на хромозома 13 (най-често присядаща анеуплоидия вследствие на грешка по време на мейозата).
- Мозайчна тризомия — само част от клетките имат допълнителното копие; клиничната картина може да бъде по‑лека и много вариабилна.
- Робертсонова транслокация — част от генетичния материал на хромозома 13 е свързан с друга хромозома (обичайно 14). Ако един от родителите е балансиран носител, детето може да наследи небалансиран вариант, което повишава риска за тризомия 13.
Клинични признаци и симптоми
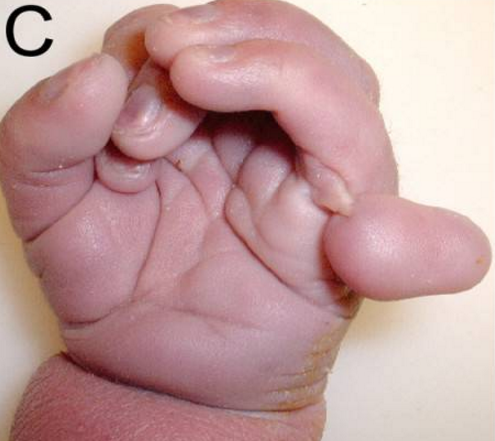
Децата със синдром на Патау имат множество вродени аномалии и тежки увреждания. Най‑често срещаните прояви включват:
- Тежка интелектуална изостаналост и забавено развитие.
- Големи вродени сърдечни пороци (напр. вентрикулен септален дефект, атриален септален дефект).
- Аномалии на лицето — цепнатини на устната и/или небцето (cleft lip/palate), малки очи (микрофталмия) или недоразвити очи.
- Наличност на допълнителни пръсти (полидактилия).
- Скалпни дефекти (cutis aplasia) — липса на кожа на част от главата.
- Холопрозенцефалия и други аномалии на мозъка.
- Нарушения в храненето, ниско тегло при раждане и затруднено дишане.
Диагностика
Диагнозата може да бъде поставена през бременността или след раждането:
- Пренатален скрининг — ултразвуковите маркери (напр. мозъчни аномалии, сърдечни дефекти, лицеви аномалии) могат да навеждат на подозрение. Неразрушаващи тестове като неинвазивен пренатален тест (NIPT / cfDNA) откриват повишен риск.
- Превантивна диагностика — потвърждава се чрез инвазивни тестове: хорионална хорионална биопсия (CVS) или амниоцентеза и последващ кариотип или молекулярни методи (FISH, микрочип, молекулярна кариотипизация).
- Постнатална диагноза — кариотип от кръвна проба потвърждава тризомия 13. Допълнителни образни и функционални изследвания (ехокардиография, невроизобразяване) се правят за оценка на уврежданията.
Лечение и грижи
Няма лечение, което да премахне генетичната причина. Подходът е симптоматичен и мултидисциплинарен:
- Поддържащо лечение и животоспасяващи интервенции при нужда (напр. хирургично коригиране на сърдечни пороци, оперативно затваряне на цепнати усти).
- Подкрепа при хранене (набутка/специални техники), управление на дихателни проблеми и инфекции.
- Реабилитация и ранна интервенция — физикална, логопедична и ерготерапия при възможност.
- Палиативна грижа и психологическа подкрепа за семейството — при тежки случаи качеството на живот и дългосрочната прогноза често налагат индивидуален подход.
Прогноза и риск
Прогнозата при тризомия 13 е сериозна. Много новородени не оцеляват дълго; преживяемостта е силно намалена, макар да има и деца с по‑дълъг живот при по‑леки форми и палиативно/специфично лечение. При мозайчната форма клиничното състояние може да бъде по‑леко и шансовете за оцеляване и по‑добро развитие са по‑големи в сравнение с класическата пълна тризомия.
Рискът детето да има тризомия 13 нараства с възрастта на майката. Ако един от родителите е балансиран носител на Робертсонова транслокация, рискът за повторение в следващи бременности е значително по‑висок и е препоръчително генетично консултиране.
Консултация и подкрепа
При подозрение или потвърдена диагноза е важно да се потърси:
- Генетичен съветник за обяснение на генетичните механизми, оценка на риска за бъдещи бременности и възможности за пренатална диагностика.
- Междуотраслов екип от педиатри, неонатолози, кардиолози, невролози, хирурзи и терапевти за координирана грижа.
- Психосоциална подкрепа за семейството и информация за местни и национални ресурси и организации, подпомагащи родители на деца с редки генетични заболявания.
Автор
AlegsaOnline.com Синдром на Патау (тризомия 13): причини, симптоми и риск Leandro Alegsa
URL: https://bg.alegsaonline.com/art/74989
Източници
- wrongdiagnosis.com : "Prevalence and Incidence of Patau syndrome"
- miscarriage.about.com : miscarriage.about.com