Тризомия 18 (Синдром на Едуардс): причини, симптоми и прогноза
Тризомия 18 (синдром на Едуардс): причини, симптоми, диагностика и прогноза — ясна информация, варианти за лечение и подкрепа за семейства.
Тризомия 18, известна още като синдром на Едуардс, е тризомия. Това е генетично заболяване. Хората с тризомия 18 имат три копия на хромозома 18. Нормалните хора имат две копия на хромозомата. Синдромът е кръстен на Джон Х. Едуардс, който го описва за първи път през 1960 г. Това е втората най-често срещана автозомна тризомия след синдрома на Даун, при която се ражда дете.
Галерия с изображения
3 Изображения

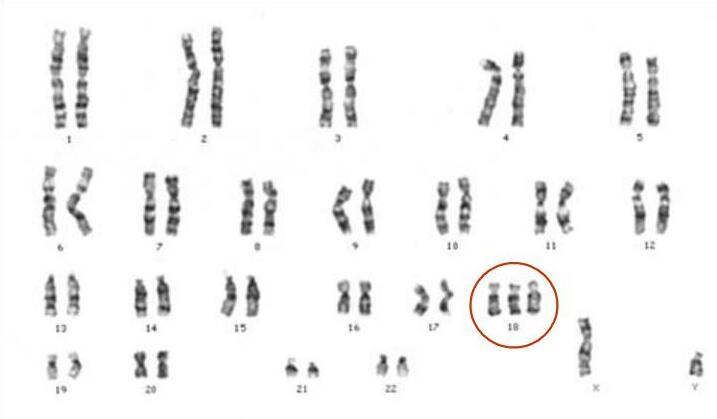
Причини и генетичен механизъм
Тризомия 18 обикновено се причинява от грешка при делене на клетките (недисджункция) по време на формиране на яйцеклетката или сперматозоида, което води до допълнително копие на хромозома 18 в оплодената яйцеклетка. Съществуват няколко форми:
- Пълна тризомия 18 — три пълни копия на хромозома 18 във всички клетки (най-често срещаната форма).
- Мозаечна тризомия 18 — допълнително копие присъства само в част от клетките; клиничната картина обикновено е по-лека. Повече за това е описано в мозаечен синдром.
- Частична тризомия или транслокация — част от хромозома 18 е допълнено върху друга хромозома (често в резултат на балансирана транслокация при един от родителите). В тези случаи рискът за повторение в следваща бременност може да бъде по-висок и се препоръчва генетично изследване на родителите.
Рискът за поява на тризомия 18 нараства с възрастта на майката. Ако в семейството има родителен балансиран транслокционен носител, рискът за повторение е различен и трябва да се обсъди с генетик.
Клинични прояви (симптоми)
Синдромът на Едуардс засяга много органи и системи. Най-честите прояви включват:
- Ниско тегло при раждане и забавен растеж (вътреутробно и постнатално).
- Микроцефалия (малка глава), характерна лицева аномалия — ниско поставени уши, малка челюст (микрогнатия), къс врат.
- Характерна поза на ръцете – с пръсти, които се застъпват (прегънати и прихванати пръсти).
- Деформирани стъпала (типично „rocker‑bottom“ стъпало), стегнати/къси крайници.
- Вродени сърдечни аномалии (напр. VSD, PDA и други), които са много чести и често определят прогнозата.
- Аномалии на бъбреците и уринарния тракт (агенезия, малформации).
- Трудности с храненето и дишането, чести инфекции, хипотония (намален мускулен тонус).
- Тежко умствено изоставане и забавено невро‑психомоторно развитие.
Диагноза
Диагнозата може да бъде поставена преди раждането или след него:
- Рутинни скрининги по време на бременността: комбинирани тестове през първия триместър (сърдечна честота, ултразвук за нухален транслуценс и биохимични маркери) и втори триместър (четворен тест). Повишен риск от тези тестове насочва към допълнителна диагностика.
- Безклетъчна фетална ДНК (cfDNA) в майчината кръв дава силен индикатор за тризомия, но не е окончателна диагноза.
- Окончателно потвърждение чрез инвазивни тестове: хорионна биопсия (CVS) или амниоцентеза с кариотипиране или молекулярна генетика.
- Ултразвукови находки, които могат да подскажат тризомия 18: вътреутробна ретардация на растежа, вродени сърдечни дефекти, омфалоцеле, кисти в хороидния плексус и характерната позиция на ръцете.
- След раждане диагнозата се потвърждава с кариотип от периферна кръв; при мозаечни форми може да е необходимо изследване на други тъкани.
Лечение и грижи
Няма лечение, което да премахне допълнителната хромозома; грижите са симптоматични и поддържащи, насочени към подобряване качеството на живот:
- Неонатална и интензивна грижа при животозастрашаващи състояния в първите дни и седмици.
- Мултидисциплинарен подход: неонатолог, кардиолог, нефролог, хирург, гастроентеролог, физиотерапевт, логопед и клиничен генетик.
- Подпомагане на храненето — често с помощта на назогастрални сонди или гастростома при проблеми с храненето.
- Хирургични корекции при определени сърдечни и други вродени аномалии — решенията са индивидуални и зависят от тежестта, общото състояние и родителските предпочитания.
- Палиативна грижа и симтоматично лечение при случаи с тежка прогноза; важно е обсъждане на целите на лечението и етичните аспекти за всяко семейство.
Прогноза и очаквано преживяване
Синдромът на Едуардс има много ниска преживяемост:
- Счита се, че около едно на 3000 живородени деца е засегнато. Честотата на заболяването се увеличава с напредване на възрастта на майката.
- Около 95% от бебетата със синдрома на Едуардс не достигат до раждане или умират преди раждане/по време на раждането. Около половината от всички бебета, родени с това заболяване, ще достигнат двумесечна възраст, а само 5–10% ще преживеят една година. Средната продължителност на живота е от пет до петнадесет дни.
- При около 1% от децата, обикновено при мозаечни и по‑леки варианти, може да се наблюдава оцеляване до детска възраст и рядко до зряла възраст, но с тежка интелектуална и физическа инвалидност.
Рискове за повторение и генетично консултиране
За повечето случаи, причинени от недисджункция, рискът за повторение в следваща бременност обикновено е нисък, но нараства с възрастта на майката. Ако при родителите бъде открита балансирана транслокация, рискът от повтаряне може да бъде значително по‑висок. Препоръчително е:
- Генетично консултиране след диагноза, за да се обсъдят причините и рисковете в бъдещи бременности.
- Предлагат се пренатални тестове в следващи бременности (скрининг и при нужда инвазивни тестове).
Подпомагане на семейството
Диагнозата на тризомия 18 е тежко емоционално предизвикателство. Подкрепата включва:
- Информация и генетично консултиране за прогнозата и възможните медицински решения.
- Психологическа подкрепа и насоки за родителите и семейството.
- Свързване с родителски и пациентски организации и групи за подкрепа, които могат да предложат практическа помощ и опит.
- Планиране на палиативни грижи и облекчение на симптомите, когато това е подходящо.
Ако вие или член на семейството сте засегнати от тризомия 18, обсъдете подробно всички въпроси с вашия акушер‑гинеколог, неонатолог или клиничен генетик — те могат да предложат най‑актуална информация, индивидуални опции за грижа и подкрепа.
Въпроси и отговори
В: Какво представлява тризомия 18?
О: Тризомия 18, известна също като синдром на Едуардс, е генетично заболяване, при което засегнатите лица имат три копия на хромозома 18 вместо нормалните две копия.
В: Кой е Джон Х. Едуардс?
О: Джон Х. Едуардс е човекът, на когото е кръстена тризомия 18. Той описва синдрома за първи път през 1960 г.
В: Колко често се среща тризомия 18?
О: Тризомия 18 засяга около едно на 3000 живородени деца.
В: Какви са някои често срещани усложнения, свързани с тризомия 18?
О: Хората с тризомия 18 често имат сърдечни аномалии, малформации на бъбреците и други нарушения на вътрешните органи.
В: Какъв е процентът на преживяемост на бебетата с тризомия 18?
О: Около 95 % от бебетата с тризомия 18 умират, преди да се родят. Около половината от всички бебета, родени с това заболяване, ще достигнат двумесечна възраст, а само 5-10% ще преживеят една година. Средната продължителност на живота е пет до петнадесет дни.
Въпрос: Каква част от децата, родени с тризомия 18, доживяват до десетгодишна възраст?
О: Един процент от децата, родени с тризомия 18, доживяват до десетгодишна възраст, обикновено в случаите на по-тежкия мозаечен синдром на Едуардс.
В: Увеличава ли се честотата на Тризомия 18 с възрастта на майката?
О: Да, честотата на Тризомия 18 се увеличава с напредването на възрастта на майката.
Свързани статии
Автор
AlegsaOnline.com Тризомия 18 (Синдром на Едуардс): причини, симптоми и прогноза Leandro Alegsa
URL: https://bg.alegsaonline.com/art/30307
Източници
- whonamedit.com : "Edwards syndrome (John Hilton Edwards)"
- nlm.nih.gov : nlm.nih.gov/MEDLINEPLUS/ency/article/001661.htm
- books.google.com : Fetal Medicine: Basic Science and Clinical Practice
- findarticles.com : findarticles.com
- emedicine.com : "Introduction to Trisomy 18"