Компютърна (изчислителна) химия — определение, методи и приложения
Компютърна химия: методи, приложения и изчислителни подходи за предсказване на структури, свойства и реактивност — от дизайна на лекарства до иновативни материали.
Компютърната химия е клон на химията, който използва инструменти и методи от компютърните науки за решаване на химични проблеми. Тези програми и алгоритми изчисляват структурата и свойствата на молекулите и на твърдите тела, като обикновено допълват или предсказват резултати, получени чрез експерименти. С помощта на изчислителни методи могат да се предвидят химични явления, които все още не са наблюдавани, и да се подпомогне проектирането на нови лекарства и материали.
Галерия с изображения
8 Изображения


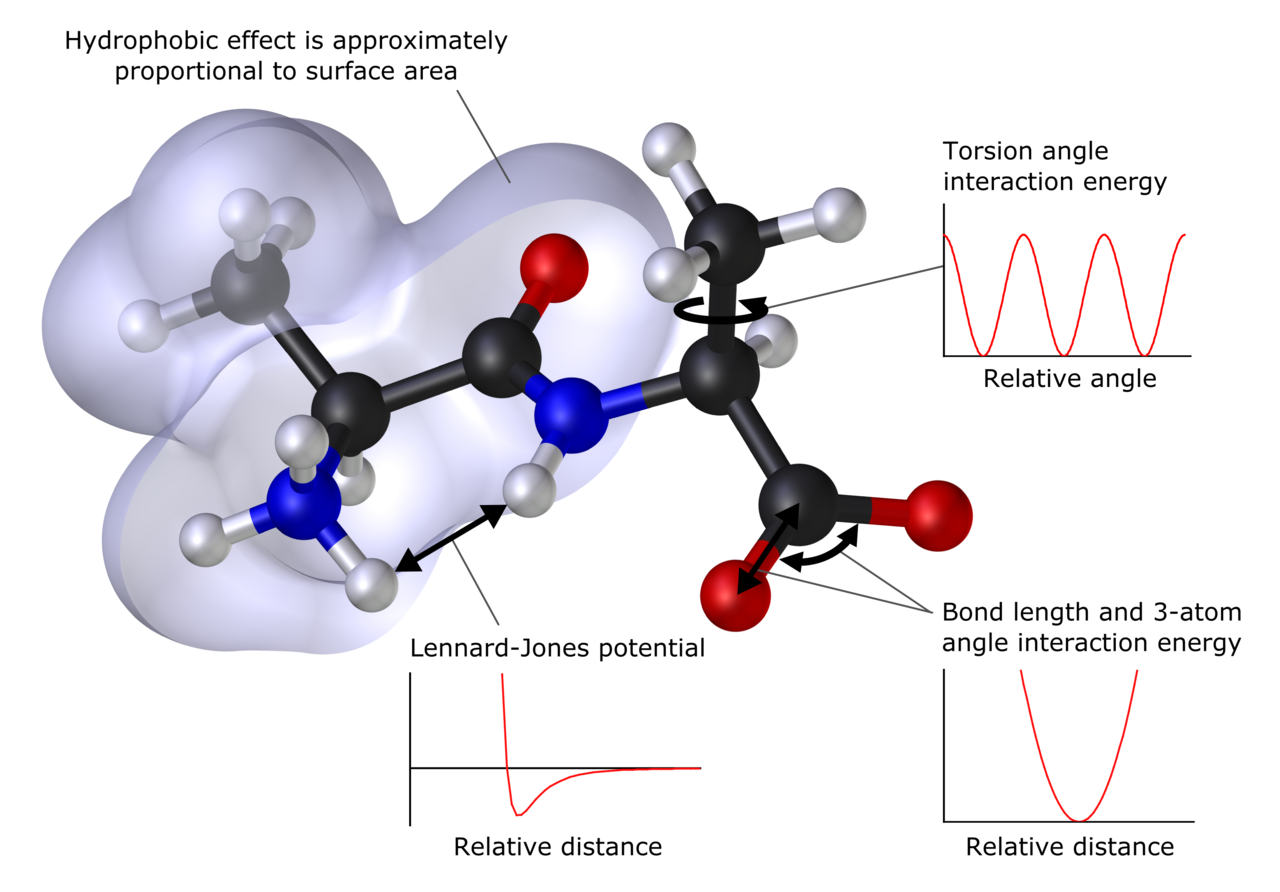


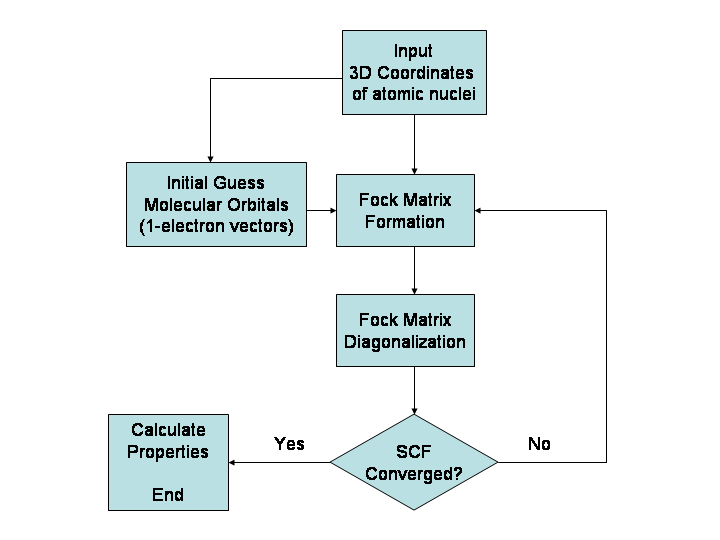
Какво може да предскаже изчислителната химия
Изчислителната химия може да предскаже:
- Структури — очакваните позиции на атомите в една молекула или в кристална решетка.
- Енергии — абсолютни и относителни енергии (на взаимодействие), бариери на реакции и стабилност на изомери.
- Електронни свойства — разпределение на електронни заряди, диполни и по-висши многополюсни моменти.
- Вибрационни честоти — нормирани честоти, които се сравняват със спектри (IR, Raman).
- Реактивност и механизми — изследване на преходни състояния и пътища на химични реакции.
- Спектроскопични величини — изчисляване на параметри за NMR, UV‑Vis, електронна структура и др.
- Сблъсъчни процеси — сечения за сблъсък с други частици и динамика на разпад.
Видове системи и скала на трудност
Изчислителната химия разглежда както статични, така и динамични системи. С нарастването на размера на изследваната система растат и изискванията към компютърното време и други ресурси (памет, дисково пространство). Тази система може да бъде единична молекула, група молекули или твърдо тяло. Методите варират от много точни до много приблизителни; високоточните методи обикновено са приложими само за малки системи.
Основни методи
- Квантово‑механични методи (ab initio) — решават уравненията на квантовата механика (например метода на Хартри–Фок и методите за корелация като MP2, CCSD). Много точни, но и много изчислително тежки.
- Плътностно‑функционална теория (DFT) — балансира точност и ресурси; широко използвана за молекули и материали. Точността зависи от избора на функционал и базисен набор.
- Полуемпирични методи — опростени квантово‑механични подходи, които използват експериментални параметри за по‑голяма бързина със загуба на точност.
- Молекулна механика (MM) — описва системите чрез класически потенциали (силови полета). Подходяща за големи биомолекули и материали; не описва прецизно химични реакции, които променят химични връзки.
- Молекулна динамика (MD) — симулира времевото развитие на системата при зададени условия (температура, налягане). Може да се комбинира с QM/MM подходи, при които реактивната област се третират квантово, а околната среда — класически.
- Монте Карло методи — използват статистическо вземане на проби за изчисляване на термодинамични свойства и равновесни състояния.
Приложения
- Проектиране на лекарства — виртуално свързване (docking), виртуален скрининг и оптимизация на лиганди спрямо целеви протеини.
- Материали и нанотехнологии — предсказване на механични, електронни и оптични свойства на нови материали, батерии, катализатори.
- Катализ и реакционни механизми — откриване на активни центрове и оптимизиране на каталитични пътища.
- Спектроскопия — подпомагане и интерпретация на експериментални спектри (IR, Raman, NMR, UV‑Vis, електронни спектри).
- Повърхностна химия — взаимодействия между молекули и повърхности, адсорбция и корозия.
- Теоретично обучение и хипотези — тестване на нови теории и модели преди експериментална проверка.
Практически аспекти и софтуер
За изчисления се използват както персонални компютри, така и високопроизводителни изчислителни клъстери и GPU ускорители. Популярни софтуерни пакети включват (примери):
- Gaussian, ORCA, GAMESS, NWChem — квантово‑химични пакети.
- VASP, Quantum ESPRESSO, CP2K — за изчисления на твърди тела и периодични системи.
- GROMACS, AMBER, CHARMM, LAMMPS — за молекулна динамика и силови полета.
- AutoDock, Schrödinger Glide — за молекулно докиране и виртуален скрининг.
Ограничения и предизвикателства
- Изчислителна сложност — някои методи имат неблагоприятно мащабиране (напр. N^4–N^7 спрямо размера на системата), което ограничава приложимостта им към големи системи.
- Приближения — всички методи използват модели и приближения (базисни набори, функционали, силови полета), които могат да въвеждат грешки.
- Солват и температурни ефекти — правилното моделиране на средата и динамиката понякога изисква комбинирани подходи и големи ресурси.
- Валидиране — резултатите често трябва да се сравнят с експериментални данни за проверка и калибрация.
Съвети за начинаещи
- Започвайте с по‑опростени методи (MM, полуемпирични подходи) за големи системи и преминавайте към DFT или ab initio за ключови вектори/реактивни центрове.
- Използвайте добре документирани базисни набори и функционали и проверявайте чувствителността на резултатите спрямо тях.
- Комбинирайте изчисления с експерименти, когато е възможно, за по‑надеждни изводи.
Изчислителната химия е мощен инструмент, който ускорява научните открития и разработката на технологии. С напредъка на хардуера и алгоритмите тя става все по‑достъпна и приложима в много области на науката и индустрията.

Свързани страници
- Биоинформатика
- Статистическа механика
Въпроси и отговори
В: Какво е изчислителна химия?
О: Изчислителната химия е клон на химията, който използва компютърните науки за решаване на химични проблеми. Тя може да се използва за изчисляване на структурата и свойствата на молекули и твърди вещества, за прогнозиране на химични явления, които все още не са наблюдавани, и за проектиране на нови лекарства и материали.
Въпрос: Какви видове системи разглежда изчислителната химия?
О: Изчислителната химия разглежда както статични, така и динамични системи. Системата може да бъде единична молекула, група молекули или твърдо тяло.
В: Какви видове информация може да предостави изчислителната химия?
О: Изчислителната химия може да предостави информация като структура (позиции на атомите), абсолютни и относителни енергии, разпределение на електронния заряд, диполи и висши многополюсни моменти, вибрационни честоти, реактивност или други спектроскопични величини, както и сечения за сблъсък с други частици.
Въпрос: Колко точни са методите, използвани в изчислителната химия?
О: Точността на методите, използвани в изчислителната химия, варира от много точна до много приблизителна. Високоточните методи обикновено са приложими само за малки системи.
В: Как изчислителната химия допълва експерименталните данни?
О: Изчислителната химия обикновено допълва информацията, получена чрез химични експерименти. Тя може да се използва за прогнозиране на резултати, които все още не са наблюдавани експериментално.
В: Размерът на изследваната система влияе ли на това колко компютърно време е необходимо?
О: Да - с нарастването на размера на изследваната система нараства и количеството компютърно време, необходимо за анализ, както и ресурсите като памет и дисково пространство, необходими за съхранение.
Свързани статии
Автор
AlegsaOnline.com Компютърна (изчислителна) химия — определение, методи и приложения Leandro Alegsa
URL: https://bg.alegsaonline.com/art/22297